
What are Thalassemias?
Thalassemias are inherited blood disorders. “Inherited” means that the disorder is passed from parents to children through genes.
Thalassemias cause the body to make fewer healthy red blood cells and less haemoglobin than normal. Haemoglobin is an iron-rich protein in red blood cells. It carries oxygen to all parts of the body. Haemoglobin also carries carbon dioxide (a waste gas) from the body to the lungs, where it’s exhaled. Low levels of haemoglobin result in mild to severe anaemia.
Causes
Haemoglobin is made of two proteins: Alpha globin and beta globin. Thalassemia occurs when there is a defect in a gene that helps control production of one of these proteins.
There are two main types of thalassemia:
*Alpha thalassemia occurs when a gene or genes related to the alpha globin protein are missing or changed (mutated).
*Beta thalassemia occurs when similar gene defects affect production of the beta globin protein
There are many forms of thalassemia. Each type has many different subtypes. Both alpha and beta thalassemia includes the following two forms:
*Thalassemia major
*Thalassemia minor
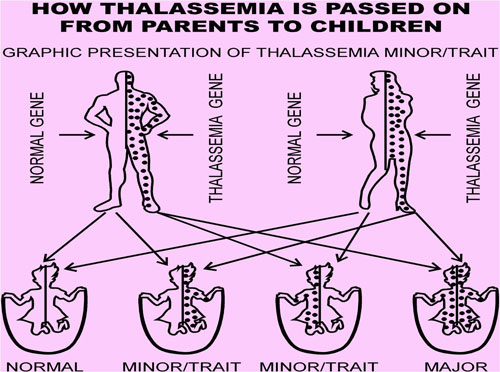
You must inherit the gene defect from both parents to develop thalassemia major.
Thalassemia minor (thalassemia trait) occurs if you receive the faulty gene from only one parent. Persons with this form of the disorder are carriers of the disease. Most of the time, they do not have symptoms.
Symptoms
Thalassemia symptoms include:
*Fatigue
*Weakness
*Pale appearance
*Yellow discoloration of skin (jaundice)
*Facial bone deformities
*Slow growth
*Abdominal swelling
*Dark urine
The signs and symptoms you experience depend on the type and severity of thalassemia you have. Some babies show signs and symptoms of thalassemia at birth, while others may develop signs or symptoms during the first two years of life. Some people who have only one affected haemoglobin gene don’t experience any thalassemia symptoms.
What are the complications of thalassemia?
Thalassemia can lead to other health problems:
•An enlarged spleen. Your spleen helps your body fight infections and filters out damaged blood cells. If you have thalassemia, your spleen may have to work harder than normal, which can cause it to enlarge. If your spleen becomes too large, it may have to be removed.
•Infections. People who have thalassemia are more likely to get blood infections, especially if they have a lot of blood transfusions. Some types of infection can be worse if you’ve had your spleen removed.
•Bone problems. Thalassemia can cause bone deformities in the face and skull. People who have thalassemia may also have severe osteoporosis (brittle bones).
•Too much iron in your blood. This can cause damage to the heart, liver, or endocrine system (glands in the body that make hormones, like the thyroid gland and adrenal glands).
Exams and Tests
Your doctor will do a physical exam to look for an enlarged spleen.
A blood sample will be sent to a laboratory to be tested.
*Red blood cells will appear small and abnormally shaped when looked at under a microscope.
*A complete blood count (CBC) reveals anaemia.
*A test called haemoglobin electrophoresis shows the presence of an abnormal form of haemoglobin.
*A test called mutational analysis can help detect alpha thalassemia
Prenatal testing
Testing can be done before a baby is born to find out if it has thalassemia and determine how severe it may be. Tests used to diagnose thalassemia in fetuses include:
•Chorionic villus sampling. This test is usually done around the 11th week of pregnancy and involves removing a tiny piece of the placenta for evaluation.
•Amniocentesis. This test is usually done around the 16th week of pregnancy and involves taking a sample of the fluid that surrounds the fetus.
Family genetic studies may help find out whether people have missing or altered haemoglobin genes that cause thalassemias

Treatment
*Treatment of thalassemia depends on the type and severity of the inherited defect. Treatments include:
*No treatment - for those with thalassemia traits and little or no anaemia
*Blood transfusion - lifelong for those with the severe homozygous form
*Chelation therapy - to manage iron overload related to frequent blood transfusions
*Management of Vitamin E and folate deficiency often associated with severe forms of thalassemia
*Stem cell transplantation - used in selected patients
*Splenectomy - may be indicated in patients with high transfusion requirements
For more information or consultation with Paediatrician Dr. Basma Atef Younes, contact Middle East Medical Center 17464848.
This blood disorder reduces the level of haemoglobin in the blood, leading to severe anaemia
Related Posts